Les causes à l’origine du développement d’une tumeur cérébrale sont encore largement inconnues. Ainsi à de très rares exceptions près, les tumeurs cérébrales ne sont pas héréditaires. De même, il n’a pas été établi de façon claire, à ce jour, de relation entre tumeurs cérébrales et des facteurs environnementaux (en particulier lignes à hautes tension, téléphones portables, pesticides…).
En revanche, les mécanismes biologiques cellulaires qui sont impliqués commencent à être bien décryptés. Les tumeurs résultent d’altérations qui concernent des gènes importants normalement présents dans l’ADN des cellules et qui jouent un rôle important dans le contrôle de la division cellulaire. Ces gènes sont susceptibles d’être altérés par exemple par des mutations qui vont pervertir leur fonction et entraîner une prolifération anarchique de la division cellulaire aboutissant à une tumeur, qui ne va cesser de croître sans aucun contrôle. Les cellules touchées par ces altérations pourraient être des cellules souches normalement présentes dans le cerveau.
Si les mécanismes intimes à l’origine des tumeurs cérébrales ciblent bien des gènes, cela n’en font pas pour autant une maladie héréditaire dans l’immense majorité des cas, car les altérations génétiques en cause sont acquises au cours de la vie et non pas transmises par l’hérédité.
Les gènes impliqués dans les tumeurs
On distingue schématiquement deux classes de gènes selon qu’ils favorisent le développement tumoral quand ils sont activés (les oncogènes) ou quand ils sont inactivés (les gènes suppresseurs de tumeurs).
A l’heure actuelle, il n’existe aucun moyen permettant d’anticiper ou d’empêcher l’apparition d’une tumeur cérébrale. Les sujets apparentés aux patients n’ont besoin d’aucune surveillance ni d’aucun dépistage. Il est important de noter qu’il n’y a aucun risque de contagion.
Oncogènes et gènes suppresseurs de tumeur
La division cellulaire est contrôlée par deux types de gènes, suivant le type de protéine qu’ils expriment. Les uns – appelés oncogènes – sont responsables de la production des effecteurs du cycle cellulaire : facteurs de croissance, récepteurs aux facteurs de croissance ou toute autre molécule qui va transmettre au noyau de la cellule un message de division cellulaire (signal mitotique). Les autres – appelés gènes suppresseurs de tumeur – sont responsables de la production de molécules qui vont au contraire bloquer la transmission de ce signal mitotique, c’est-à-dire de division cellulaire. Une division cellulaire normale peut ainsi être vue comme un équilibre délicat entre l’action des oncogènes (accélérateur de la division) et des gènes suppresseurs de tumeur (frein à la division). La croissance tumorale résulte en grande partie du déséquilibre entre ces deux types d’effecteurs, par inactivation des deux copies chromosomiques d’un gène suppresseur de tumeur, ou par activation d’un oncogène.
L’inhibition de la transmission du signal constitue un objectif majeur de la recherche sur les tumeurs au cerveau.
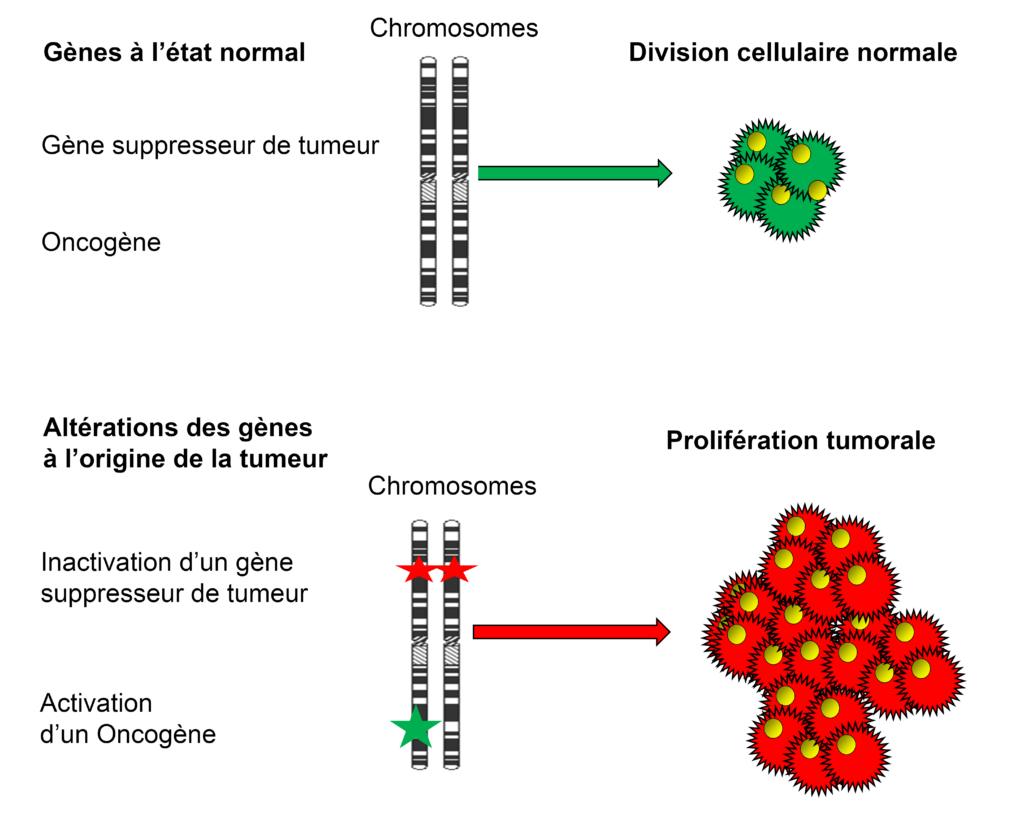
On connaît maintenant un bon nombre des altérations génétiques impliquées dans les tumeurs cérébrales. L’enchaînement de ces altérations génétiques est à l’origine de la progression tumorale vers la malignité, Certains gènes étant altérés de façon précoce, d’autres intervenant plus tardivement au cours de l’évolution. En outre, les altérations génétiques diffèrent suivant le type de tumeur considérée, par exemple entre tumeurs astrocytaires et oligodendrogliales.
Il est ainsi possible aujourd’hui, à partir de l’étude de l’ADN tumoral, de dresser une carte d’identité génétique de la tumeur : on parle désormais de diagnostic histo-moléculaire des tumeurs. D’autre part l’identification de ces altérations causales dans la génèse des tumeurs permet d’élaborer des stratégies thérapeutiques ciblées.
Les voies de progression des gliomes
L’identification d’altérations génétiques impliquées dans la génèse des gliomes permet donc une nouvelle classification à la fois histologique et moléculaire, appelée le « diagnostic intégré » (2).Ces altérations peuvent concerner :
- Une inactivation de gènes suppresseurs de tumeur (identifiés comme p53, p16, PTEN, CIC ou FUBP1 ou non identifiés mais localisés sur les bras chromosomiques 1p et 19q),
- Une activation d’oncogènes (comme l’EGFR ou TERT),
- Une mutation dans un gène impliqué dans le métabolisme, en modifiant la fonction (comme la mutation des gènes IDH1 ou IDH2). Elles sont associées à une hyperméthylation de l’ADN appelée « phénotype méthylateur des ilots CpG » qui bloquerait la différenciation cellulaire,
- Une mutation dans un gène impliqué dans l’expression des autres gènes comme une mutation des gènes histones H3.
On distingue ainsi, désormais, 3 voies principales de développement des gliomes (3) :
- La voie dépendante de la mutation IDH. Celle-ci est observée chez 70% des gliomes de grade II et III et 85% des glioblastomes secondaires (faisant suite à l’évolution d’un gliome de grade II ou III). Associée à la codélétion 1p19q, elle définit le diagnostic d’oligodendrogliome IDH muté codélété 1p19q, d’évolution lente et particulierement sensible au traitement de radiothérapie et de chimiothérapie. En absence de codélétion 1p19q, on retient le diagnostic d’astrocytome IDH muté ou de glioblastome IDH muté
- La voie dite IDH Wild-type (ou IDH « sauvage »), caractérisée par l’absence de mutation IDH et de mutation des histones H3, aboutissant le plus souvent au diagnostic de glioblastome
- La voie des mutations des histones H3, jamais associée à une mutation du gène IDH et surtout observé dans les gliomes du sujet jeune ou dans les gliomes dits de la ligne médiane (axe central du système nerveux).
Ces différentes altérations moléculaires sont désormais recherchées de façon courante et ce, dès le diagnostic. Cela nécessite le recours à des plateformes dites de biologie moléculaire ou les différentes analyses nécessaires seront réalisées après extraction de l’ADN tumoral à partir des cellules tumorales. La réalisation de cette caractérisation moléculaire peut prendre quelques semaines.
Elle est cependant indispensable au diagnostic et à la décision de traitement. La meilleure compréhension des mécanismes biologiques impliqués dans la glioma-gènése ouvre aussi la perspective de thérapies ciblées, adaptées au profil moléculaire de chaque tumeur et ainsi, à une médecine personnalisée.

Figure 2: Classification histo-moléculaire des gliomes (selon OMS 2016) ou diagnostic intégré
Références scientifiques :
- Epidemiology for primary brain tumors: a nationwide population-based study. Darlix A, Zouaoui S, Rigau V, Bessaoud F, Figarella-Branger D, Mathieu-Daudé H, Trétarre B, Bauchet F, Duffau H, Taillandier L, Bauchet L.J Neurooncol. 2017 Feb;131(3):525-546.
- Louis DN, Perry A, Reifenberger G, von Deimling A, Figarella-Branger D, Cavenee WK, Ohgaki H, Wiestler OD, Kleihues P, Ellison DW. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: a summary. Acta Neuropathol. 2016 Jun;131(6):803-20.
- Gliomes diffus de l’adulte. Bielle F, Figarella-Branger D, Mokhtari K. La lettre du neurologue 2016 juin XX : 158-162